
The Bottom Line: “Adult Onset Still’s Disease (AOSD) is a rare systemic inflammatory disorder characterized by daily fever, inflammatory polyarthritis, and a transient salmon-pink maculopapular rash. AOSD is alternatively known as systemic onset juvenile idiopathic arthritis. Even though there is no specific diagnostic test, a serum ferritin level more than 1000ng/ml is common in AOSD.”
Category Archives: Kokko Conference
Grady Kokko conference: In patients less than 45 years old, what is multiple myeloma’s (MM’s) incidence, presentation, and prognosis?
Bottom line: Less than 4 percent of new cases of multiple myeloma (MM) occur in patients less than 45 years old.(1) A study has found that younger patients aged 21-40 had higher incidences of lytic lesions and high-risk cytogenetic abnormalities but a “lower rate of elevated lactate dehyrdrogenase.”(2) Two studies within the last five years suggest that the prognosis for young patients with stage 1 MM “may be as good if not better than in the general population of MM patients.”(2,3)
References:
1. DynaMed Plus [Internet]. Ipswich (MA): EBSCO Information Services. 1995 – . Record No. T116744, Multiple myeloma; [updated 2019 Oct 1, cited 2019 Oct 14]. Emory login required.
2. Jurczyszyn A, Nahi H, Avivi I, et al. Characteristics and outcomes of patients with multiple myeloma aged 21-40 years versus 41-60 years: a multi-institutional case-control study. British Journal of Haematology. 2016 Dec;175(5):884-891. doi: 10.1111/bjh.14328.
3. Jurczyszyn A, Davila J, Kortüm KM, et al. Multiple myeloma in patients up to 30 years of age: a multicenter retrospective study of 52 cases. Leukemia & Lymphoma. 2019 Feb;60(2):471-476. doi: 10.1080/10428194.2018.1480766.
4. National Cancer Institute Surveillance, Epidemiology, and End Results (SEER) Program. Cancer Stat Facts: Myeloma. Accessed October 14, 2019.
Summary:
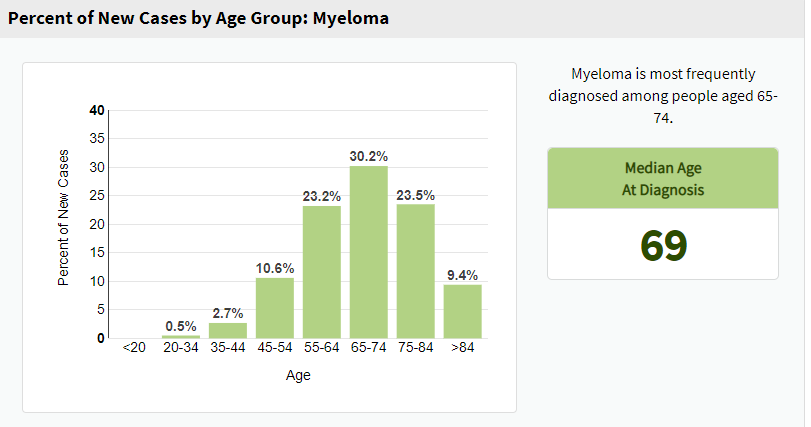 (4)
(4)
Age-adjusted incidence by age
36.3 per 100,000 persons in adults ≥ 65 years old
2.7 per 100,000 persons in patients < 65 years old (1)
In a case-control study of 173 patients aged 21-40 and 916 patients aged 41-60 in the novel agent era between 2000 and 2015, “younger patients presented with a higher incidence of lytic lesions (82% vs. 72%; P = 0·04) and high-risk cytogenetic abnormalities (83% vs. 68%; P = 0·007), but lower rate of elevated lactate dehydrogenase (21% vs. 44%; P < 0·001). Five- and 10-year overall survival (OS) in younger versus older patients was 83% vs. 67% and 56% vs. 39%, respectively (P < 0·001). Similar results were seen when studying the subset of 780 patients who underwent autologous transplantation. Younger patients with ISS stage 1 had a better OS than older patients (P < 0·001). There was no survival difference between younger and older patients with ISS stage 2 or 3.”(2)
In a study of 52 patients diagnosed with MM aged 8-30 years old, “68% of patients had International Scoring System (ISS) 1 MM; 22% presented with the light chain-only disease, and 48% with elevated serum lactate dehydrogenase (LDH)….The group was followed-up for the median period of 86 months. The median overall survival (OS) was 166 months (95% CI: 53-222), with 5-year OS rate of 77% (95% CI: 61.0-87.9).”(3)
Grady Kokko conference: Review of granulomatous amebic encephalitis (GAE)
Bottom line: GAE’s symptoms mimic meningitis and/or brain abscesses, and a positive CSF culture is needed to confirm the diagnosis. Treatment usually consists of Miltefosine along with other medication(s). Most cases are fatal.
Reference: 1. Pana A, Anilkumar AC. Amebic meningoencephalitis. StatPearls;2019 January 11.
Summary: GAE is a very rare condition caused by ubiquitous amaeoba that are found in fresh water, lakes, and rivers; it strikes both immunocompromised and immunocompetent individuals. It has symptoms that closely resemble those of meningitis or a brain abscess. A culture of cerebrospinal fluid (CSF) is needed to confirm the diagnosis. The two pathways of GAE are through the cornea to the central nervous system (CNS) or, more commonly, from the lungs or skin to the CNS. Many parts of the upper spinal cord and/or brain may be affected simultaneously; thus. Imaging findings vary. CT may show pseudotumoral lesions, largely isolated lesions, progressive hydrocephalus, meningeal thickening, and/or multifocal ring-enhancing lesions. MRI may show edema, multiple ring-enhancing lesions, and multifocal lesions. These nonspecific findings may suggest either primary amebic meningoenceophalitis or GAE. Drug combinations and varying regimens are used to treat GAE. Miltefosine is usually given along with one or more of the following: Sulfadiazine, Pentamidine, Trimethoprim/sulfamethoxazole, Amphotericin, Flucytosine, and Fluconazole or the related drugs itraconazole or voriconazole. The unusual symptomology and need for a CSF culture result in its mortality rate of above 90%.1
Grady Kokko Conference: What are the most common causes of Loeffler’s endocarditis?
The Bottom Line: Cardiac involvement in hypereosinophilic syndrome (HES) is termed Loeffler’s endocarditis. Reported secondary causes of HES include parasitic infections (including helminths), adverse drug reactions, allergies, leukemia, lymphomas, some other solid tumors, and vasculitis (most commonly eosinophilic granulomatosis with polyangitis (formerly Churg-Strauss).(1-2)
References:
- Hussain N, Patel P, Yin J, Davis R, Ikladios O. A case of Loeffler’s endocarditis after initiation of adalimumab. J Hosp Intern Med Perspect. 2019 Feb 11;9(1):29-32. doi:doi: 10.1080/20009666.2018.1562852. PMID: 30788072.</li.
- Goldstein JA. Chapter 24: Restrictive cardiomyopathy. In: Crawford MH, ed. Current Diagnosis & Treatment: Cardiology. 5th ed. McGraw-Hill Education; 2017.
Summary: A rare blood disorder, HES results in dysregulation and overproduction of eosinophils, resulting in end-organ damage. The most common cause of morbidity and mortality in HES patients is cardiac involvement, termed Loeffler’s endocarditis.““`
Grady Kokko Conference: Review of Epstein-Barr virus (EBV)-associated hemophagocytic lymphohistiocytosis (HLH)
The Bottom Line: Infection-associated HLH is most commonly caused by EBV.1 Distinguishing between routine EBV infection and HLH complicating EBV infection is very difficult but can be done by looking at timing and severity of manifestations.2 The HLH-1994 and HLH-2004 protocols guide treatment, but other regimens have been used recently.1
References:
-
Lai W, Wang Y, Wang J, Wu L, Jin Z, Wang Z. Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in adults and adolescents—a life-threatening disease: analysis of 133 cases from a single center. Hematology. 2018 Dec;23(10):810-816. Doi:10.1080/10245332.2018.
-
Marsh RA. Epstein-Barr virus and hemophagocytic lymphohistiocytosis. Frontiers in Immunology. 2018 Jan 8;8:1902. Doi:10.3389/fimmu.2017.01902.
Summary: Most patients with routine EBV infections do not appear toxic. In contrast, those with EBV-associated HLH usually appear ill; generally they have intense fevers that persist, life-threatening reductions in red blood cells, a need for supportive transfusions, a clotting defect, and profound central nervous system involvement. Altered mental status, seizures, focal or global deficits, and acute liver failure that necessitates a transplant may occur. The HLH-94/2004 regimen calls for etoposide, dexamethasone, and cyclosporine.2 In one study of 133 adolescents and adults with EBV-associated HLH, 112 received the aforementioned regimen, and 46% of those responded partially or completely. Five of six who received L-DEP regimen responded (regimen consisted of liposomal doxorubicin together with etoposide and highdose methylprednisolone in combination with PEG-aspargase). Five of 15 who were given initial treatment without etoposide achieved a partial response. Refractory or relapsed patients received L-DEP or DEP regimen (liposomal doxorubicin together with etoposide and highdose methylprednisolone), and 79.71% of 69 patients responded partially or completely. Thirty-six of 133 patients received stem cell transplants, and their overall survival rate was 52.8%. 78% of the study’s 133 patients died within one year.1
